
this review was composed by Andreas Gewies in 2003
The following paragraphs will introduce you to the morphological and molecular features of apoptotic cell death and will discuss the involvement of apoptosis in physiological and pathological processes.
pathways
pathways
apoptosis
pathways
go to Index
Already since the mid-nineteenth century, many observations have indicated that cell death plays a considerable role during physiological processes of multicellular organisms, particularly during embryogenesis and metamorphosis [Gluecksmann, 1951; Lockshin, 2001]. The term programmed cell death was introduced in 1964, proposing that cell death during development is not of accidential nature but follows a sequence of controlled steps leading to locally and temporally defined self-destruction [Lockshin, 1964].
Eventually, the term apoptosis had been coined in order to describe the morphological processes leading to controlled cellular self-destruction and was first introduced in a publication by Kerr, Wyllie and Currie [Kerr, 1972]. Apoptosis is of greek origin, having the meaning "falling off or dropping off", in analogy to leaves falling off trees or petals dropping off flowers. This analogy emphasizes that the death of living matter is an integral and necessary part of the life cycle of organisms. The apoptotic mode of cell death is an active and defined process which plays an important role in the development of multicellular organisms and in the regulation and maintenance of the cell populations in tissues upon physiological and pathological conditions. It should be stressed that apoptosis is a well-defined and possibly the most frequent form of programmed cell death, but that other, non-apoptotic types of cell death also might be of biological significance [Leist, 2001a].
go to Index
The development and maintenance of multicellular biological systems depends on a sophisticated interplay between the cells forming the organism, it sometimes even seems to involve an altruistic behaviour of individual cells in favour of the organism as a whole. During development many cells are produced in excess which eventually undergo programmed cell death and thereby contribute to sculpturing many organs and tissues [see Figure 1 and Meier, 2000].

A particularly instructive example for the implication of programmed cell death in animal development is the formation of free and independent digits by massive cell death in the interdigital mesenchymal tissue [Zuzarte-Luis, 2002]. Other examples are the development of the brain, during which half of the neurons that are initially created will die in later stages when the adult brain is formed [Hutchins, 1998] and the development of the reproductive organs [Meier, 2000]. Also cells of an adult organism constantly undergo physiological cell death which must be balanced with proliferation in order to maintain homeostasis in terms of constant cell numbers. The majority of the developing lymphocytes die either during genetic rearrangement events in the formation of the antigen receptor, during negative selection or in the periphery, thereby tightly controlling the pool of highly efficient and functional but not self-reactive immune cells and at the same time keeping lymphocyte numbers relatively constant [Rathmell, 2002].
Taken together, apoptotic processes are of widespread biological significance, being involved in e.g. development, differentiation, proliferation/homoeostasis, regulation and function of the immune system and in the removal of defect and therefore harmful cells. Thus, dysfunction or dysregulation of the apoptotic program is implicated in a variety of pathological conditions. Defects in apoptosis can result in cancer, autoimmune diseases and spreading of viral infections, while neurodegenerative disorders, AIDS and ischaemic diseases are caused or enhanced by excessive apoptosis [Fadeel, 1999a].
Due to its importance in such various biological processes, programmed cell death is a widespread phenomenon, occuring in all kinds of metazoans [Tittel, 2000] such as in mammals, insects [Richardson, 2002], nematodes [Liu, 1999b], and cnidaria [Cikala, 1999]. Moreover, programmed cell death also might play a role in plant biology [Solomon, 1999], and apoptosis-like cell death mechanisms even have been observed and used as a model system in yeast [Frohlich, 2000; Skulachev, 2002]. Fascinating insights into the origin and evolution of programmed cell death might possibly be given by the fact, that programmed cell death is also an integral part of the life cycle of other unicellular eukaryotes (such as the kinetoplastid parasite Trypanosoma brucei brucei, the ciliate Tetrahymena thermophila, and the slime mold Dictyostelium discoideum) and that even prokaryotes (such as Bacillus subtilis, Streptomyces and Myxobacteria) sometimes undergo regulated cell death [Ameisen, 2002].
go to Index
Apoptotic cells can be recognized by stereotypical morphological changes: the cell shrinks, shows deformation and looses contact to its neighbouring cells. Its chromatin condenses and marginates at the nuclear membrane, the plasma membrane is blebbing or budding, and finally the cell is fragmented into compact membrane-enclosed structures, called 'apoptotic bodies' which contain cytosol, the condensed chromatin, and organelles (Figure 2). The apoptotic bodies are engulfed by macrophages and thus are removed from the tissue without causing an inflammatory response. Those morphological changes are a consequence of characteristic molecular and biochemical events occurring in an apoptotic cell, most notably the activation of proteolytic enzymes which eventually mediate the cleavage of DNA into oligonucleosomal fragments as well as the cleavage of a multitude of specific protein substrates which usually determine the integrity and shape of the cytoplasm or organelles [Saraste, 2000].
Apoptosis is in contrast to the necrotic mode of cell-death in which case the cells suffer a major insult, resulting in a loss of membrane integrity, swelling and disrupture of the cells. During necrosis, the cellular contents are released uncontrolled into the cell's environment which results in damage of surrounding cells and a strong inflammatory response in the corresponding tissue [Leist, 2001a].
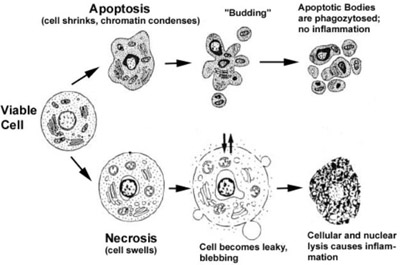
go to Index
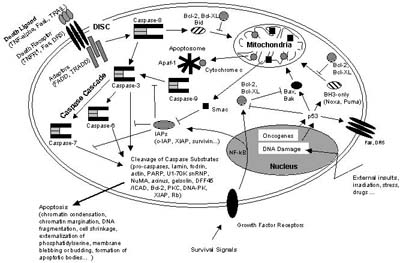
This paragraph is meant to provide a general overview of basic apoptotic signalling pathways and of the molecular machinery responsible for the induction and execution of apoptosis. The most important signalling molecules and cellular structures will be discussed in context of their function and of the mechanisms in which they are involved in the initiation, mediation, execution, and regulation of apoptosis. This chapter should give an impression of the sophisticated interplay between factors that promote or suppress apoptosis, resulting in a complicated regulatory network which determines the fate of an individual cell as part of its multicellular environment (Figure 6).
go to Index
Apoptosis is a tightly regulated and at the same time highly efficient cell death program which requires the interplay of a multitude of factors. The components of the apoptotic signalling network are genetically encoded and are considered to be usually in place in a nucleated cell ready to be activated by a death-inducing stimulus [Ishizaki, 1995; Weil, 1996]. Apoptosis can be triggered by various stimuli from outside or inside the cell, e.g. by ligation of cell surface receptors, by DNA damage as a cause of defects in DNA repair mechanisms, treatment with cytotoxic drugs or irradiation, by a lack of survival signals, contradictory cell cycle signalling or by developmental death signals. Death signals of such diverse origin nevertheless appear to eventually activate a common cell death machinery leading to the characteristic features of apoptotic cell death.
Much of the understanding of cell death has come from genetic studies in the nematode C. elegans by which several genes have been identified that function in the apoptotic killing and elimination of 131 of the initially 1090 somatic cells that are generated druing hermaphrodite development [Hengartner, 1999]. The proximal cause of apoptosis in C. elegans is the activation of the cysteine protease ced-3, which is mediated by its oligomerization at the activator protein ced-4. Activity of the ced-3/ced-4 complex is regulated by the apoptosis inhibitor ced-9 and the apoptosis inducer egl-1 (Figure 3). Subsequent studies in mammals and in the fly, D. melanogaster, have identidied counterparts for these C. elegans genes, demonstrating that the core components of the cell death machinery are conserved through evolution [Richardson, 2002]. Accordingly, ced-3 is the single C. elegans member of a family of cysteine proteases, the caspases, whereas ced-4 corresponds to the mammalian apoptotic protease activating factor 1, Apaf-1, which is the core of a caspase-activating signalling complex, the apoptosome. Egl-1 and ced-9 are members of the Bcl-2 family of pro- or antiapoptotic proteins, respectively, which play an important role in the mediation and regulation of apoptotic signalling pathways. All of those central components will be discussed within the following paragraphs.
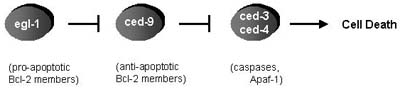
go to Index
The caspases, cysteine proteases homologous to C. elegans ced-3, are of central importance in the apoptotic signalling network which are activated in most cases of apoptotic cell death [Bratton, 2000]. Actually, strictly defined, cell death only can be classified to follow a classical apoptotic mode if execution of cell death is dependent on caspase activity [Leist, 2001a].
The term caspases is derived from cysteine-dependent aspartate-specific proteases: their catalytical activity depends on a critical cysteine-residue within a highly conserved active-site pentapeptide QACRG, and the caspases specifically cleave their substrates after Asp residues. So far, 7 different caspases have been identified in Drosophila, and 14 different members of the caspase-family have been described in mammals, with caspase-11 and caspase�12 only identified in the mouse [Denault, 2002; Richardson, 2002]. According to a unified nomenclature, the caspases are referred to in the order of their publication: caspase-1 is ICE (Interleukin-1�-Converting Enzyme), the first mammalian caspase described to be a homologue of Ced-3 [Creagh, 2001; Miura, 1993]. Caspase-1 as well as caspases-4, -5, -11, and �12 appear to be mainly involved in the proteolytic maturation of pro-inflammatory cytokines such as pro-IL-1� and pro-IL-18 and their contribution to the execution of apoptosis remains questionable [Denault, 2002]. Indeed, mice deficient for caspase-1 or caspase-11 develop normally and cells from those knockout mice remain sensitive to various death stimuli [Li, 1995; Wang, 1998]. In contrast, gene knockout experiments targeting caspase-3 and caspase-9 resulted in perinatal mortality as a result of severe defects in brain development [Kuida, 1998; Kuida, 1996], whereas caspase-8 deficient embryos died after day 12 [Varfolomeev, 1998]. This and the observation that cell lines derived from those knockout experiments are resistant to distinct apoptosis stimuli underlines the importance of caspases as proapoptotic mediators. Indeed, caspase-3, caspase-9, caspase-8, and additionally caspases-2, -6, -7, and �10 have been recognized to play an important role in the apoptotic signalling machinery [Earnshaw, 1999].
In the cell, caspases are synthesized as inactive zymogens, the so called procaspases, which at their N-terminus carry a prodomain followed by a large and a small subunit which sometimes are separated by a linker peptide. Upon maturation, the procaspases are proteolytically processed between the large and small subunit, resulting in a small and a large subunit. The prodomain is also frequently but not necessarily removed during the activation process. A heterotetramer consisting of each two small and two large subunits then forms an active caspase. The proapoptotic caspases can be divided into the group of initiator caspases including procaspases-2, -8, -9 and �10, and into the group of executioner caspases including procaspases-3, -6, and �7. Whereas the executioner caspases possess only short prodomains, the initiator caspases possess long prodomains, containing death effector domains (DED) in the case of procaspases-8 and �10 or caspase recruitment domains (CARD) as in the case of procaspase-9 and procaspase-2. Via their prodomains, the initiator caspases are recruited to and activated at death inducing signalling complexes either in response to the ligation of cell surface death receptors (extrinsic apoptosis pathways) or in response to signals originating from inside the cell (intrinsic apoptosis pathways).
In extrinsic apoptosis pathways (Figure 4), e.g. procaspase-8 is recruited by its DEDs to the death inducing signalling complex (DISC), a membrane receptor complex formed following to the ligation of a member of the tumor necrosis factor receptor (TNFR) family [Sartorius, 2001]. When bound to the DISC, several procaspase-8 molecules are in close proximity to each other and therefore are assumed to activate each other by autoproteolysis [Denault, 2002].
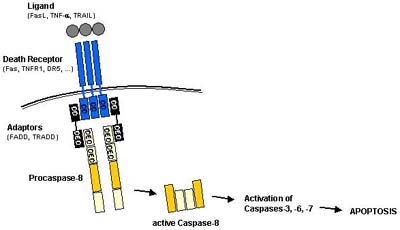
Intrinsic apoptosis pathways (Figure 5) involve procaspase-9 which is activated downstream of mitochondrial proapoptotic events at the so called apoptosome, a cytosolic death signalling protein complex that is formed upon release of cytochrome c from the mitochondria [Salvesen, 2002b]. In this case it is the dimerization of procaspase-9 molecules at the Apaf-1 scaffold that is responsible for caspase-9 activation [Denault, 2002]. Once the initiator caspases have been activated, they can proteolytically activate the effector procaspases-3, -6, and -7 which subsequently cleave a specific set of protein substrates, including procaspases themselves, resulting in the mediation and amplification of the death signal and eventually in the execution of cell death with all the morphological and biochemical features usually observed [Earnshaw, 1999].
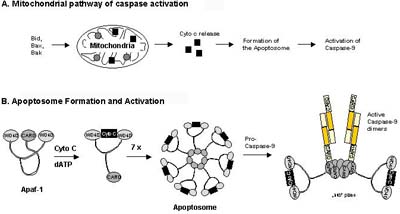
go to Index
Extrinsic apoptosis signalling is mediated by the activation of so called �death receptors� which are cell surface receptors that transmit apoptotic signals after ligation with specific ligands. Death receptors belong to the tumor necrosis factor receptor (TNFR) gene superfamily, including TNFR-1, Fas/CD95, and the TRAIL receptors DR-4 and DR-5 [Ashkenazi, 2002]. All members of the TNFR family consist of cysteine rich extracellular subdomains which allow them to recognize their ligands with specificity, resulting in the trimerization and activation of the respective death receptor [Naismith, 1998]. Subsequent signalling is mediated by the cytoplasmic part of the death receptor which contains a conserved sequence termed the death domain (DD). Adapter molecules like FADD or TRADD themselves possess their own DDs by which they are recruited to the DDs of the activated death receptor, thereby forming the so-called death inducing signalling complex (DISC). In addition to its DD, the adaptor FADD also contains a death effector domain (DED) which through homotypic DED-DED interaction sequesters procaspase-8 to the DISC (Figure 4). As described above, the local concentration of several procaspase-8 molecules at the DISC leads to their autocatalytic activation and release of active caspase-8. Active caspase-8 then processes downstream effector caspases which subsequently cleave specific substrates resulting in cell death. Cells harboring the capacity to induce such direct and mainly caspase-dependent apoptosis pathways were classified to belong to the so called type I cells [Scaffidi, 1998].
In type II cells, the signal coming from the activated receptor does not generate a caspase signalling cascade strong enough for execution of cell death on its own. In this case, the signal needs to be amplified via mitochondria-dependent apoptotic pathways. The link between the caspase signalling cascade and the mitochondria is provided by the Bcl-2 family member Bid. Bid is cleaved by caspase-8 and in its truncated form (tBID) translocates to the mitochondria where it acts in concert with the proapoptotic Bcl-2 family members Bax and Bak to induce the release of cytochrome c and other mitochondrial proapoptotic factors into the cytosol (see Figure 6 and [Luo, 1998]). Cytosolic cytochrome c is binding to monomeric Apaf-1 which then, in a dATP-dependent conformational change, oligomerizes to assemble the apoptosome, a complex of wheel-like structure with 7-fold symmetry, that triggers the activation of the initiator procaspase-9 (see Figure 5 and [Acehan, 2002]). Activated caspase-9 subsequently initiates a caspase cascade involving downstream effector caspases such as caspase-3, caspase-7, and caspase-6, ultimately resulting in cell death [Slee, 1999].
go to Index
Besides amplifying and mediating extrinsic apoptotic pathways, mitochondria also play a central role in the integration and propagation of death signals originating from inside the cell such as DNA damage, oxidative stress, starvation, as well as those induced by chemotherapeutic drugs [Kaufmann, 2000; Wang, 2001]. Most apoptosis-inducing conditions involve the disruption of the mitochondrial inner transmembrane potential (Dy) as well as the so called permeability transition (PT), a sudden increase of the inner mitochondrial membrane permeability to solutes with a molecular mass below approximately 1.5 kDa. Concomitantly, osmotic mitochondrial swelling has been observed by influx of water into the matrix with eventual rupture of the outer mitochondrial membrane, resulting in the release of proapoptotic proteins from the mitochondrial intermembrane space into the cytoplasm [Bernardi, 1999; Loeffler, 2000]. Released proteins include cytochrome c, which activates the apoptosome and therefore the caspase cascade, but also other factors such as the apoptosis-inducing factor AIF [Susin, 1999], the endonuclease endoG [Li, 2001], Smac/Diablo [Verhagen, 2000], and Htr/Omi [Verhagen, 2002]. Interestingly, PT is always followed by Dy, but Dy is not always caused by PT, and cytochrome c release has been observed even in absence of Dy [Bernardi, 1999; Kroemer, 2000]. In addition to the release of mitochondrial factors, the dissipation of Dy and PT also cause a loss of the biochemical homeostasis of the cell: ATP synthesis is stopped, redox molecules such as NADH, NADPH, and glutathione are oxidized, and reactive oxygen species (ROS) are increasingly generated [Kroemer, 2000; Kroemer, 1997]. Increased levels of ROS directly cause the oxidation of lipids, proteins, and nucleic acids, thereby enhancing the disruption of Dy as part of a positive feedback [Marchetti, 1997]. Several possible mechanisms for PT have been proposed, but there appears to exist consent that a so-called permeability transition pore (PTP) is formed consisting of the adenin nucleotide translocator (ANT) and the voltage-dependent anion channel (VDAC) as its core components. ANT is the most abundant protein of the inner mitochondrial membrane and as a transmembrane channel is responsible for the export of ATP in exchange with ADP (antiport). Overexpression of ANT-1 in human cancer cell lines dominantly induces apoptosis with all its characteristic features whereas its closely conserved homologue ANT-2 does not, indicating a specific mechanistic role of ANT-1 in mitochondrial apoptosis events [Bauer, 1999]. VDAC, also called porin, is the most abundant protein of the outer mitochondrial membrane and forms a non-selective pore through the outer membrane. Indicated by direct protein-protein interactions, VDAC-ANT complexes presumably connect inner and outer mitochondrial membrane to so-called �contact sites�, corresponding to a close association of the two membranes and thereby possibly constituting the PT pore [Beutner, 1998]. Since PT, loss of Dy, and release of mitochondrial proteins are of central importance in mediating and enhancing apoptotic pathways, those mitochondrial events must be kept under strict control of regulatory mechanisms which are in many ways dependent on members of the Bcl-2 family which will be discussed in the next paragraph.
go to Index
Commonly, the activation of apoptosis is regarded to occur when a cell encounters a specific death-inducing signal such as the ligation of a death receptor by its cognate ligand or if cells are treated with a cytotoxic drug. This suggests that the apoptosis signalling pathways in viable cells are kept in an inactive state and are only turned on in response to a death stimulus. But it should be taken into account that the components of the apoptotic signalling network are genetically encoded and ready for action in most cell types. Therefore, an interesting and possibly more realistic alternative view would be as follows: all cells of a multicellular animal might be intrinsically programmed to self-destruct and indeed would die instantaneously unless cell death is continously repressed by survival signals such as provided by other cells of the organism, e.g. growth factors, hormones, nutrients. Those survival signals enhance the expression and/or activity of antiapoptotic regulatory molecules thereby keeping in check the activation of proapoptotic factors [Ameisen, 2002; Raff, 1993]. Indeed, a set of various antiapoptotic molecules and mechanisms has been identified, as well as proapoptotic factors that counteract those inhibitory molecules when apoptotic demise of a cell is timely and imperative.
go to Index
Bcl-2, an oncogene which in follicular lymphoma is frequently linked to an immunoglobulin locus by the chromosome translocation t(14:18), was the first example of an oncogene that inhibits cell death rather than promoting proliferation. B cells transfected with Bcl-2 were shown to be rendered resistant towards apoptosis induced by IL-3 withdrawal: for the first time it was shown that the pathway toward tumorigenesis depends not only on the ability to escape growth control but also depends on the ability to prevent apoptosis [Vaux, 1988].
When homologues of Bcl-2 had been identified, it became apparent that a Bcl-2 family of proteins can be defined by the presence of conserved sequence motifs known as Bcl-2 homology domains (BH1 to BH4). In mammals, up to 30 relatives have been decribed of which some belong to a group of pro-survival members and others to a group of proapoptotic members [Borner, 2003]. In addition to Bcl-2 itself, there are a number of other prosurvival proteins, e.g. Bcl-XL, Bcl-w, A1, and Mcl-1, which all possess the domains BH1, BH2, BH3, and BH4. The proapoptotic group of Bcl-2 members can be devided into two subgroups: the Bax-subfamily consists of Bax, Bak, and Bok that all possess the domains BH1, BH2, and BH3, whereas the BH3-only proteins (Bid, Bim, Bik, Bad, Bmf, Hrk, Noxa, Puma, Blk, BNIP3, and Spike) have only the short BH3 motif, an interaction domain that is both necessary and sufficient for their killing action [Cory, 2002; Mund, 2003].
There has been quite some debate about how the Bcl-2 family controls apoptosis: one model proposes that Bcl-2 members might directly control caspase activation [Strasser, 2000], whereas another model claims that they mainly act by guarding mitochondrial integrity [Wang, 2001]. In support of the first model, the worm Bcl-2 orthologue ced-9 binds to the Apaf-1-like adaptor protein ced-4 and prevents it from activating the caspase ced-3 unless the BH3-only protein Egl-1 displaces ced-4 as shown in Figure 3 [Conradt, 1998]. In contrast, the mammalian ced-4 homologue Apaf-1 obviously does not interact with Bcl-2-like proteins [Moriishi, 1999] but is activated by cytosolic cytochrome c (see Figure 5), and it is the release of cytochrome c from the mitochondria that can be controlled by Bcl-2 [Kluck, 1997; Yang, 1997]. Therefore it appears likely, that the central function of mammalian Bcl-2 family members is to guard mitochondrial integrity and to control the release of mitochondrial proteins into the cytoplasm [Cory, 2002].
How then is mitochondrial integrity affected by proapoptotic Bcl-2 family members? Central to this question are Bax and Bak, even though inactivation of the Bax gene alone affected apoptosis only slightly and disruption of Bak alone did not show any effect. However, the double knockout of Bax and Bak resulted in dramatic impairment of apoptosis during development in many tissues with superfluous cells accumulating in the hematopoietic system and in the brain. Additionally, cells derived from those Bax -/- Bak -/- mice are insensitive to treatment with e.g. etoposide or irradiation [Lindsten, 2000; Wei, 2001]. Bax is a cytosolic monomer in viable cells but during apoptosis changes its conformation, integrates into the outer mitochondrial membrane and oligomerizes [Nechushtan, 2001]. Although the mechanism is controversial, Bax and Bak oligomers are believed to provoke or contribute to the permeabilization of the outer mitochondrial membrane (PT), either by forming channels by themselves [Antonsson, 2000] or by interacting with components of the PT pore such as VDAC [Tsujimoto, 2000].
In contrast, antiapoptotic Bcl-2 members sequester proapoptotic Bcl-2 members by bindig to their BH3 domains and thereby ultimately prevent Bax or Bak activation/ oligomerization and consequently inhibit mitochondrial proapoptotic events: overexpression of Bcl-2 or Bcl-XL potently inhibits apoptosis in response to many cytotoxic insults, among others by suppressing the generation of ROS, stabilizing Dy, preventing PT and consequently blocking the release of e.g. cytochrome c [Reed, 1998]. Besides eliciting its antiapoptotic effects on the mitochondrial level by indirectly controlling the activation of the apoptosome, Bcl-2 also appears to inhibit apoptotic pathways that are independent of Apaf-1/caspase-9 and which might depend on caspase-7 as a central effector [Marsden, 2002]. In this context one might even expect the existence of another but up to now unidentified Apaf-1 homologue that can be directly controlled by Bcl-2/Bcl-XL [Puthalakath, 2002].
Whereas Bax and Bak represent the central core of a proapoptotic Bcl-2 death machinery that is held in check by the pro-survival members Bcl-2 and Bcl-XL, members of the BH3-only subfamily are required for the activation of proapoptotic Bax/Bak function [Bouillet, 2002]. On the other hand, the killing effect of BH3-only members depends on Bax/Bak, since cells double-deficient for Bax and Bak do not die upon overexpression of BH3-only proteins as it would be the case in wildtype cells, indicating that BH3-only members function upstream of Bax and Bak [Lindsten, 2000; Wei, 2001]. Importantly, just as the proapoptotic activity of multidomain proteins Bax and Bak is controlled by their interaction with the antiapoptotic guardians Bcl-2/Bcl-XL, also most BH3-only members display a strong binding preference to antiapoptotic Bcl-2/Bcl-XL and in this way are kept under control [Scorrano, 2003].
Individual BH3-only proteins are believed to transduce specific death signals since they can be activated for apoptosis signalling by sensing cell stress, such as DNA damage (Noxa and Puma are p53-inducible genes), growth factor deprivation (Hrk and Bim mRNA expression is increased), or anoikis (Bmf is activated by subcellular relocalization) [Borner, 2003]. As another example, cytoplasmic Bid is processed by caspase-8 to its truncated form tBid, which after myristoylation translocates to the mitochondria where it triggers cytochrome c release by affecting Bax/Bak oligomerization and/or by mobilizing cytochrome c stores in cristae [Cory, 2002; Scorrano, 2002].
In general, BH3-only proteins are thought to interfere with the fine-tuned balance of homo- or heterooligomerization between proapoptotic multidomain members Bax/Bak and antiapoptotic members Bcl-2/Bcl-XL (Figure 7). It has been proposed that Bid and Bim possess BH3 domains (Bid-like BH3 domain) which can directly mediate Bax/Bak oligomerization, whereas Bad and Bik possess Bad-like BH3 domains which do not directly act on Bax/Bak but preferentially interact with antiapoptotic Bcl-2/Bcl-XL. As a consequence, activated Bad/Bik might be able to displace Bid/Bim from the binding pocket of antiapoptotic Bcl-2/Bcl-XL, and - in this way released - Bid/Bim might provoke Bax/Bak oligomerization and cytochrome c release even at subliminal levels [Letai, 2002].
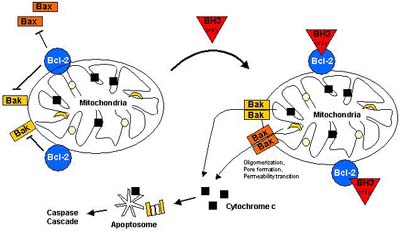
In summary, a current model of how Bcl-2 family members regulate apoptosis can be descibed as follows (Figure 7): specific apoptotic
stress signals trigger the activation of particular BH3-only proteins which then interact with antiapoptotic members on the outer mitochondrial
(but also nuclear/ER) membrane, resulting in the release of Bax-like proapoptotic factors. Bax-like factors undergo a conformational change
(possibly assisted by some BH3-only proteins), insert into the outer mitochondrial membrane where they provoke PT and the release of
apoptogenic factors [Borner, 2003]
go to Index
Expression levels of antiapoptotic proteins such as Bcl-2, Bcl-XL, and A1 were reported to be upregulated by the transcription factor NF-kB which besides being a central regulator of the innate and adaptive immune response is commonly described as an antiapoptotic transcription factor [Heckman, 2002; Karin, 2002], although under certain circumstances NF-kB also might positively contribute to apoptosis induction [Grimm, 1996b]. Besides inducing the expression of pro-survival Bcl-2 members, NF-kB additionally transactivates a number of other antiapoptotic genes, such as the IAPs (inhibitors of apoptosis proteins).
IAPs are a family of antiapoptotic proteins whose prototype originally was described in baculovirus with many homologues found to be conserved across several species. So far, eight human IAP homologues have been identified, among others NAIP, c-IAP1, c-IAP2, XIAP and survivin. All IAPs contain baculovirus IAP repeat (BIR) domains, 70 amino acid motifs, which are essential for the antiapoptotic properties of IAPs [Takahashi, 1998] because it is the interaction between the BIR domains and caspases that is believed to confer most of the antiapoptotic activity of IAPs. Indeed, XIAP, c-IAP1 and c-IAP2 are thought to directly inhibit caspases-3, -7, and �9 [Salvesen, 2002a]. In case of XIAP, it is the BIR3 domain that directly binds to the small subunit of caspase-9, whereas it is the BIR2 domain that interacts with the active-site substrate binding pocket of caspases-3 and �7 [Huang, 2001; Srinivasula, 2001]. In addition to the BIR domains, c-IAP1, c-IAP2, and XIAP contain a highly conserved RING domain at their C-terminal end which possesses E3 ubiquitin ligase activity. Via this RING domain, IAPs are able to catalyze their own ubiquitination, thereby targeting themselves for degradation by the proteasome [Yang, 2000], but they also might target other proteins such as caspase-3 and �7 for ubiquitination and degradation [Huang, 2000; Suzuki, 2001]. Direct inhibition of caspase activity by c-IAPs is certainly a very important means of regulation when considered that signalling cascades mediated by proteolytic enzymes such as caspases is irreversible once activated and therefore must be precisely regulated in order to prevent locally and temporally inappropriate demise of cells. Importantly, Smac/Diablo, when released from the mitochondrial intermembrane space during mitochondrial apoptotic events, is able to counteract the inhibitory effect of IAPs on caspases since Smac/Diablo can bind to e.g. XIAP in a manner that displaces caspases from XIAP and enables their activation. Thus, Smac/Diablo is a negative regulator of IAPs and in this way unfolds its apoptosis-enhancing property [Du, 2000]. Essential for the ability of Smac/Diablo to bind to XIAP and to release caspases is a conserved tetrapeptide motif which is also present in HtrA2/Omi, another mitochondrial proapoptotic factor [van Loo, 2002], as well as in the Drosophila proapoptotic proteins Reaper, Hid, Grim, and Sickle which can bind to DIAP1, a Drosophila IAP and thereby support activation of the fly caspase Dronc [Hawkins, 2000].
go to Index
In the adult human body several hundred thousand cells are produced every second by mitosis, and a similar number die by apoptosis for the maintenance of homeostasis and for specific tasks such as the regulation of immune cell selection and activity [Fadeel, 1999b]. Dysregulation of apoptotic signalling can play a primary or secondary role in various diseases with insufficient apoptosis leading to e.g. cancer (cell acumulation, resistance to therapy, defective tumor surveillance by the immune system), autoimmunity (failure to eliminate autoreactive lymphocytes), persistent infections (failure to eradicate infected cells), whereas excessive apoptosis contributes to e.g. neurodegeneration (Alzheimers� disease, Parkinson�s disease, Huntington�s disease, amyotrophic lateral sclerosis), autoimmunity (uncontrolled apoptosis induction in specific organs), AIDS (depletion of T lymphocytes), and ischaemia (stroke, myocardial infarction) [Reed, 2002]. Malfunction of the death machinery results from the mutation of genes that code for factors directly or indirectly involved in the initiation, mediation, or execution of apoptosis, and several mutations in apoptosis genes have been identified as a causing or contributing factor in human diseases [Mullauer, 2001].
Of special interest is the involvement of defective apoptosis pathways in tumor formation, progression, and metastasis as well as the occurrence of multidrug resistance during cancer therapy [Johnstone, 2002]. During the last years it became more and more evident that tumorigenesis is not merely the result of excessive proliferation due to the activation of oncogenes but to the same extent depends on the � frequently concurrent - impairment of apoptosis checkpoints [Hanahan, 2000; Wang, 1999]. Intriguingly, many of the alterations that induce malignant transformation, such as oncogene-driven deregulated proliferation and invasion, actually sensitize a cell to apoptosis, and therefore only those oncogenic transformed cells will survive and become malignant which additionally acquire defects in apoptosis pathways and therefore are protected against cell death induction [Vousden, 2002]. A transformed cell can achieve protection against apoptosis by inappropriate activation or expression of antiapoptotic proteins (which usually act as oncogenes), or by the inactivation of proapoptotic factors (which usually are tumor-suppressors).
As an example and as already mentioned, Bcl-2 was the first apoptosis-related gene that was recognized to play a role in tumorigenesis, and indeed, Bcl-2 is overexpressed in a variety of cancers, contributing to cancer cell survival through direct inhibition of apoptosis [Hockenbery, 1990; Reed, 1999]. Conversely, mutated or downregulated Bax and Bak are observed in certain cancers [Kondo, 2000; Rampino, 1997] and disruption of those genes promotes tumorigenesis in mice [Yin, 1997]. Further, proapoptotic Bad and procaspase-9 are negatively regulated by the oncogenic Akt/PKB kinase, which on the other hand is frequently constitutively active or amplified in many types of human cancer [Nicholson, 2002], and its antagonist, the phosphatase PTEN, is one of the most commonly mutated tumor-suppressors [Yamada, 2001]. Even more underlining its potential as an oncogene, Akt/PKB is also stimulating the NF-kB survival pathway by phosphorylation of IkB kinase alpha (IKK alpha) and it is suppressing p53 proapoptotic signalling by phosphorylation of the oncogene Mdm2 which thereby is activated for inhibition of p53 [Mayo, 2002]. Both, NF-kB and Mdm2, are themselves inappropriately activated or overexpressed in the process of transformation [Chene, 2003; Orlowski, 2002].
A paradigm for the central importance of apoptosis checkpoints in the defense against malignant transformation presents the tumor suppressor p53, which is presumably the most intensely studied apoptosis factor contributing to cancer because it is inactivated in presumably more than 50% of all human cancers [Hainaut, 2000]. p53 is a tumor suppressor protein which is activated as a transcription factor in response to e.g. oncogene activation, hypoxia and especially DNA damage, resulting in growth arrest and/or apoptosis by stimulating the expression of various p53 target genes such as p21, Bax, Puma, Noxa, Apaf-1, Fas, and DR5 [Vousden, 2002] or by repressing the expression of antiapoptotic proteins, e.g. Bcl-2, Bcl-XL or survivin [Hoffman, 2002; Wu, 2001]. Recent evidence suggests transcription-independent p53 apoptosis pathways in which p53 translocates to the mitochondria, interacts with Bcl-XL, induces PT and the release of cytochrome c [Mihara, 2003]. In non-stressed, undamaged cells p53 therefore must be kept under stringent control: it is present only at low cellular concentrations, it is retained in the cytosol and prevented to enter the nucleus, and its transactivation domain is inactivated [Chene, 2003]. Central to p53 regulation is the oncogene Mdm2 which binds to and thereby inhibits p53 (see Figure 8). Mdm2 is a ubiquitin-ligase which mediates ubiquitination of p53, thereby targeting it for degradation by the proteasome. In this way, p53 levels are kept low in normal cells [Kubbutat, 1997]. The importance of Mdm2 in the control of p53 is demonstrated by mdm2 gene knockout mice which die early during development but are rescued from death by additional deletion of the p53 gene [Montes de Oca Luna, 1995].
In response to cellular stress (such as DNA damage), p53 is phosphorylated at specific serine/threonine residues which prevents the Mdm2-p53 interaction, and thus p53 is stabilized and activated [Schon, 2002]. Moreover, p53 is central to oncogene-induced cell death because it is induced by oncogenes such as c-myc, adenovirus E1A, and ras as well as by loss of the retinoblastoma tumor suppressor pRb [Henriksson, 2001]. All those oncogenes activate the transcription factor E2F-1 which not only can promote cell cycle progression and proliferation but at the same time directly triggers expression of the tumor suppressor ARF which leads to stabilization and activation of p53 [Ginsberg, 2002]. This explains in part why oncogene activation not always leads to uncontrolled cell proliferation but under certain circumstances to the stabilization of p53 and activation of cell death, provided that p53 signalling pathways are intact [Eischen, 1999].
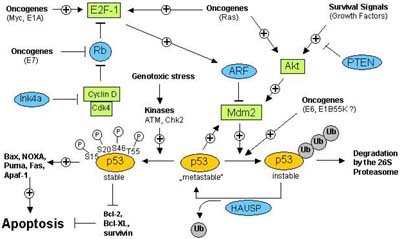
Therefore in many instances, an oncogenic insult only results in increased proliferation and eventually malignant transformation if activators of p53 (such as ARF, Chk2, or ATM), p53 itself (e.g. by Mdm2, the adenovirus E1B, papillomavirus E6, or SV40 large T antigen), or p53 downstream signalling components (p53 target genes) are inactivated (Figure 8). On the other hand, p53-mediated apoptosis pathways can be suppressed by survival signals, such as growth factors binding to their cognate growth factor receptors what eventually results in activation of the Akt kinase [Datta, 1999]. Akt kinase is known to mediate a number of antiapoptotic mechanisms, such as the direct phosphorylation and inactivation of Bad and caspase-9, the activation of NF-kB antiapoptotic signalling via phosphorylation of IkB, but also phoshorylation and activation of Mdm2 as an inhibitor of p53 [Mayo, 2002]. Besides phosphorylation, ubiquitination and protein-protein interactions, p53 is also regulated by acetylation what affects its transcriptional activity, as well as by sumoylation [Melchior, 2003; Appella, 2001].
Gaining insight into the mechanisms and alterations by which components of the apoptotic machinery contribute to pathogenic processes, should allow the development of more effective, higher specific and therefore better-tolerable therapeutic approaches. Those may include the targeted activation of proapoptotic tumor suppressors or alternatively the blockade of antiapoptotic oncogenes in the case of cancer, whereas for the treatment of premature cell death during e.g. neurodegeneration the inhibition of proapoptotic key components such as the caspases might be promising [Reed, 2002].
go to Index
Acehan, D, Jiang, X, Morgan, DG, Heuser, JE, Wang, X and Akey, CW (2002). "Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation." Mol Cell 9(2): 423-32.
Ameisen, JC (2002). "On the origin, evolution, and nature of programmed cell death: a timeline of four billion years." Cell Death Differ 9(4): 367-93.
Antonsson, B, Montessuit, S, Lauper, S, Eskes, R and Martinou, JC (2000). "Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria." Biochem J 345(Pt 2): 271-8.
Appella, E (2001). "Modulation of p53 function in cellular regulation." Eur J Biochem 268(10): 2763.
Ashkenazi, A (2002). "Targeting death and decoy receptors of the tumour-necrosis factor superfamily." Nat Rev Cancer 2(6): 420-30.
Bauer, MK, Schubert, A, Rocks, O and Grimm, S (1999). "Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis." J Cell Biol 147(7): 1493-502.
Bernardi, P, Scorrano, L, Colonna, R, Petronilli, V and Di Lisa, F (1999). "Mitochondria and cell death. Mechanistic aspects and methodological issues." Eur J Biochem 264(3): 687-701.
Beutner, G, Ruck, A, Riede, B and Brdiczka, D (1998). "Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases." Biochim Biophys Acta 1368(1): 7-18.
Borner, C (2003). "The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions." Mol Immunol 39(11): 615-47.
Bouillet, P and Strasser, A (2002). "BH3-only proteins - evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death." J Cell Sci 115(Pt 8): 1567-74.
Bratton, SB, MacFarlane, M, Cain, K and Cohen, GM (2000). "Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis." Exp Cell Res 256(1): 27-33.
Chene, P (2003). "Inhibiting the p53-MDM2 interaction: an important target for cancer therapy." Nat Rev Cancer 3(2): 102-9.
Cikala, M, Wilm, B, Hobmayer, E, Bottger, A and David, CN (1999). "Identification of caspases and apoptosis in the simple metazoan Hydra." Curr Biol 9(17): 959-62.
Conradt, B and Horvitz, HR (1998). "The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9." Cell 93(4): 519-29.
Cory, S and Adams, JM (2002). "The Bcl2 family: regulators of the cellular life-or-death switch." Nat Rev Cancer 2(9): 647-56.
Creagh, EM and Martin, SJ (2001). "Caspases: cellular demolition experts." Biochem Soc Trans 29(Pt 6): 696-702.
Datta, SR, Brunet, A and Greenberg, ME (1999). "Cellular survival: a play in three Akts." Genes Dev 13(22): 2905-27.
Denault, JB and Salvesen, GS (2002). "Caspases: keys in the ignition of cell death." Chem Rev 102(12): 4489-500.
Du, C, Fang, M, Li, Y, Li, L and Wang, X (2000). "Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition." Cell 102(1): 33-42.
Earnshaw, WC, Martins, LM and Kaufmann, SH (1999). "Mammalian caspases: structure, activation, substrates, and functions during apoptosis." Annu Rev Biochem 68: 383-424.
Eischen, CM, Weber, JD, Roussel, MF, Sherr, CJ and Cleveland, JL (1999). "Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis." Genes Dev 13(20): 2658-69.
Fadeel, B, Gleiss, B, Hogstrand, K, Chandra, J, Wiedmer, T, Sims, PJ, Henter, JI, Orrenius, S and Samali, A (1999a). "Phosphatidylserine exposure during apoptosis is a cell-type-specific event and does not correlate with plasma membrane phospholipid scramblase expression." Biochem Biophys Res Commun 266(2): 504-11.
Fadeel, B, Orrenius, S and Zhivotovsky, B (1999b). "Apoptosis in human disease: a new skin for the old ceremony?" Biochem Biophys Res Commun 266(3): 699-717.
Frohlich, KU and Madeo, F (2000). "Apoptosis in yeast--a monocellular organism exhibits altruistic behaviour." FEBS Lett 473(1): 6-9.
Ginsberg, D (2002). "E2F1 pathways to apoptosis." FEBS Lett 529(1): 122-5.
Gluecksmann, A (1951). "Cell death in normal vertebrate ontogeny." Biological Reviews 26: 59-86.
Grimm, S, Bauer, MK, Baeuerle, PA and Schulze-Osthoff, K (1996). "Bcl-2 down-regulates the activity of transcription factor NF-kappaB induced upon apoptosis." J Cell Biol 134(1): 13-23.
Hainaut, P and Hollstein, M (2000). "p53 and human cancer: the first ten thousand mutations." Adv Cancer Res 77: 81-137.
Hanahan, D and Weinberg, RA (2000). "The hallmarks of cancer." Cell 100(1): 57-70.
Hawkins, CJ, Yoo, SJ, Peterson, EP, Wang, SL, Vernooy, SY and Hay, BA (2000). "The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM." J Biol Chem 275(35): 27084-93.
Heckman, CA, Mehew, JW and Boxer, LM (2002). "NF-kappaB activates Bcl-2 expression in t(14;18) lymphoma cells." Oncogene 21(24): 3898-908.
Hengartner, MO (1999). "Programmed cell death in the nematode C. elegans." Recent Prog Horm Res 54: 213-22; discussion 222-4.
Henriksson, M, Selivanova, G, Lindstrom, M and Wiman, KG (2001). "Inactivation of Myc-induced p53-dependent apoptosis in human tumors." Apoptosis 6(1-2): 133-7.
Hockenbery, D, Nunez, G, Milliman, C, Schreiber, RD and Korsmeyer, SJ (1990). "Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death." Nature 348(6299): 334-6.
Hoffman, WH, Biade, S, Zilfou, JT, Chen, J and Murphy, M (2002). "Transcriptional repression of the anti-apoptotic survivin gene by wild type p53." J Biol Chem 277(5): 3247-57.
Huang, H, Joazeiro, CA, Bonfoco, E, Kamada, S, Leverson, JD and Hunter, T (2000). "The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7." J Biol Chem 275(35): 26661-4.
Huang, Y, Park, YC, Rich, RL, Segal, D, Myszka, DG and Wu, H (2001). "Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain." Cell 104(5): 781-90.
Hutchins, JB and Barger, SW (1998). "Why neurons die: cell death in the nervous system." Anat Rec 253(3): 79-90.
Ishizaki, Y, Cheng, L, Mudge, AW and Raff, MC (1995). "Programmed cell death by default in embryonic cells, fibroblasts, and cancer cells." Mol Biol Cell 6(11): 1443-58.
Johnstone, RW, Ruefli, AA and Lowe, SW (2002). "Apoptosis: a link between cancer genetics and chemotherapy." Cell 108(2): 153-64.
Karin, M and Lin, A (2002). "NF-kappaB at the crossroads of life and death." Nat Immunol 3(3): 221-7.
Kaufmann, SH and Earnshaw, WC (2000). "Induction of apoptosis by cancer chemotherapy." Exp Cell Res 256(1): 42-9.
Kerr, JF, Wyllie, AH and Currie, AR (1972). "Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics." Br J Cancer 26(4): 239-57.
Kluck, RM, Bossy-Wetzel, E, Green, DR and Newmeyer, DD (1997). "The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis." Science 275(5303): 1132-6.
Kondo, S, Shinomura, Y, Miyazaki, Y, Kiyohara, T, Tsutsui, S, Kitamura, S, Nagasawa, Y, Nakahara, M, Kanayama, S and Matsuzawa, Y (2000). "Mutations of the bak gene in human gastric and colorectal cancers." Cancer Res 60(16): 4328-30.
Kroemer, G and Reed, JC (2000). "Mitochondrial control of cell death." Nat Med 6(5): 513-9.
Kroemer, G, Zamzami, N and Susin, SA (1997). "Mitochondrial control of apoptosis." Immunol Today 18(1): 44-51.
Kubbutat, MH, Jones, SN and Vousden, KH (1997). "Regulation of p53 stability by Mdm2." Nature 387(6630): 299-303.
Kuida, K, Haydar, TF, Kuan, CY, Gu, Y, Taya, C, Karasuyama, H, Su, MS, Rakic, P and Flavell, RA (1998). "Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9." Cell 94(3): 325-37.
Kuida, K, Zheng, TS, Na, S, Kuan, C, Yang, D, Karasuyama, H, Rakic, P and Flavell, RA (1996). "Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice." Nature 384(6607): 368-72.
Leist, M and Jaattela, M (2001). "Four deaths and a funeral: from caspases to alternative mechanisms." Nat. Rev. Mol. Cell Biol. 2(8): 589-98.
Letai, A, Bassik, MC, Walensky, LD, Sorcinelli, MD, Weiler, S and Korsmeyer, SJ (2002). "Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics." Cancer Cell 2(3): 183-92.
Li, LY, Luo, X and Wang, X (2001). "Endonuclease G is an apoptotic DNase when released from mitochondria." Nature 412(6842): 95-9.
Li, P, Allen, H, Banerjee, S, Franklin, S, Herzog, L, Johnston, C, McDowell, J, Paskind, M, Rodman, L, Salfeld, J and et al. (1995). "Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock." Cell 80(3): 401-11.
Lindsten, T, Ross, AJ, King, A, Zong, WX, Rathmell, JC, Shiels, HA, Ulrich, E, Waymire, KG, Mahar, P, Frauwirth, K, Chen, Y, Wei, M, Eng, VM, Adelman, DM, Simon, MC, Ma, A, Golden, JA, Evan, G, Korsmeyer, SJ, MacGregor, GR and Thompson, CB (2000). "The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues." Mol Cell 6(6): 1389-99.
Liu, QA and Hengartner, MO (1999). "The molecular mechanism of programmed cell death in C. elegans." Ann N Y Acad Sci 887: 92-104.
Lockshin, RA and Williams, CM (1964). "Programmed cell death. II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths." J Insect Physiol 10: 643-649.
Lockshin, RA and Zakeri, Z (2001). "Programmed cell death and apoptosis: origins of the theory." Nat Rev Mol Cell Biol 2(7): 545-50.
Loeffler, M and Kroemer, G (2000). "The mitochondrion in cell death control: certainties and incognita." Exp Cell Res 256(1): 19-26.
Luo, X, Budihardjo, I, Zou, H, Slaughter, C and Wang, X (1998). "Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors." Cell 94(4): 481-90.
Marchetti, P, Decaudin, D, Macho, A, Zamzami, N, Hirsch, T, Susin, SA and Kroemer, G (1997). "Redox regulation of apoptosis: impact of thiol oxidation status on mitochondrial function." Eur J Immunol 27(1): 289-96.
Marsden, VS, O'Connor, L, O'Reilly, LA, Silke, J, Metcalf, D, Ekert, PG, Huang, DC, Cecconi, F, Kuida, K, Tomaselli, KJ, Roy, S, Nicholson, DW, Vaux, DL, Bouillet, P, Adams, JM and Strasser, A (2002). "Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome." Nature 419(6907): 634-7.
Mayo, LD and Donner, DB (2002). "The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network." Trends Biochem Sci 27(9): 462-7.
Meier, P, Finch, A and Evan, G (2000). "Apoptosis in development." Nature 407(6805): 796-801.
Melchior, F and Hengst, L (2002). "SUMO-1 and p53." Cell Cycle 1(4): 245-9.
Mihara, M, Erster, S, Zaika, A, Petrenko, O, Chittenden, T, Pancoska, P and Moll, UM (2003). "p53 has a direct apoptogenic role at the mitochondria." Mol Cell 11(3): 577-90.
Miura, M, Zhu, H, Rotello, R, Hartwieg, EA and Yuan, J (1993). "Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3." Cell 75(4): 653-60.
Montes de Oca Luna, R, Wagner, DS and Lozano, G (1995). "Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53." Nature 378(6553): 203-6.
Moriishi, K, Huang, DC, Cory, S and Adams, JM (1999). "Bcl-2 family members do not inhibit apoptosis by binding the caspase activator Apaf-1." Proc Natl Acad Sci U S A 96(17): 9683-8.
Mullauer, L, Gruber, P, Sebinger, D, Buch, J, Wohlfart, S and Chott, A (2001). "Mutations in apoptosis genes: a pathogenetic factor for human disease." Mutat Res 488(3): 211-31.
Mund, T, Gewies, A, Schoenfeld, N, Bauer, MK and Grimm, S (2003). "Spike, a novel BH3-only protein, regulates apoptosis at the endoplasmic reticulum." Faseb J 17(6): 696-8.
Naismith, JH and Sprang, SR (1998). "Modularity in the TNF-receptor family." Trends Biochem Sci 23(2): 74-9.
Nechushtan, A, Smith, CL, Lamensdorf, I, Yoon, SH and Youle, RJ (2001). "Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis." J Cell Biol 153(6): 1265-76.
Nicholson, KM and Anderson, NG (2002). "The protein kinase B/Akt signalling pathway in human malignancy." Cell Signal 14(5): 381-95.
Orlowski, RZ and Baldwin, AS, Jr. (2002). "NF-kappaB as a therapeutic target in cancer." Trends Mol Med 8(8): 385-9.
Puthalakath, H and Strasser, A (2002). "Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins." Cell Death Differ 9(5): 505-12.
Raff, MC, Barres, BA, Burne, JF, Coles, HS, Ishizaki, Y and Jacobson, MD (1993). "Programmed cell death and the control of cell survival: lessons from the nervous system." Science 262(5134): 695-700.
Rampino, N, Yamamoto, H, Ionov, Y, Li, Y, Sawai, H, Reed, JC and Perucho, M (1997). "Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype." Science 275(5302): 967-9.
Rathmell, JC and Thompson, CB (2002). "Pathways of apoptosis in lymphocyte development, homeostasis, and disease." Cell 109(Suppl): S97-107.
Reed, JC (2002). "Apoptosis-based therapies." Nat Rev Drug Discov 1(2): 111-21.
Reed, JC (1998). "Bcl-2 family proteins." Oncogene 17(25): 3225-36.
Reed, JC (1999). "Dysregulation of apoptosis in cancer." J Clin Oncol 17(9): 2941-53.
Richardson, H and Kumar, S (2002). "Death to flies: Drosophila as a model system to study programmed cell death." J Immunol Methods 265(1-2): 21-38.
Salvesen, GS and Duckett, CS (2002a). "IAP proteins: blocking the road to death's door." Nat Rev Mol Cell Biol 3(6): 401-10.
Salvesen, GS and Renatus, M (2002b). "Apoptosome: the seven-spoked death machine." Dev Cell 2(3): 256-7.
Saraste, A and Pulkki, K (2000). "Morphologic and biochemical hallmarks of apoptosis." Cardiovasc Res 45(3): 528-37.
Sartorius, U, Schmitz, I and Krammer, PH (2001). "Molecular mechanisms of death-receptor-mediated apoptosis." Chembiochem 2(1): 20-9.
Scaffidi, C, Fulda, S, Srinivasan, A, Friesen, C, Li, F, Tomaselli, KJ, Debatin, KM, Krammer, PH and Peter, ME (1998). "Two CD95 (APO-1/Fas) signaling pathways." Embo J 17(6): 1675-87.
Schon, O, Friedler, A, Bycroft, M, Freund, SM and Fersht, AR (2002). "Molecular mechanism of the interaction between MDM2 and p53." J Mol Biol 323(3): 491-501.
Scorrano, L, Ashiya, M, Buttle, K, Weiler, S, Oakes, SA, Mannella, CA and Korsmeyer, SJ (2002). "A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis." Dev Cell 2(1): 55-67.
Scorrano, L and Korsmeyer, SJ (2003). "Mechanisms of cytochrome c release by proapoptotic BCL-2 family members." Biochem Biophys Res Commun 304(3): 437-44.
Skulachev, VP (2002). "Programmed death in yeast as adaptation?" FEBS Lett 528(1-3): 23-6.
Slee, EA, Harte, MT, Kluck, RM, Wolf, BB, Casiano, CA, Newmeyer, DD, Wang, HG, Reed, JC, Nicholson, DW, Alnemri, ES, Green, DR and Martin, SJ (1999). "Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner." J Cell Biol 144(2): 281-92.
Solomon, M, Belenghi, B, Delledonne, M, Menachem, E and Levine, A (1999). "The involvement of cysteine proteases and protease inhibitor genes in the regulation of programmed cell death in plants." Plant Cell 11(3): 431-44.
Srinivasula, SM, Hegde, R, Saleh, A, Datta, P, Shiozaki, E, Chai, J, Lee, RA, Robbins, PD, Fernandes-Alnemri, T, Shi, Y and Alnemri, ES (2001). "A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis." Nature 410(6824): 112-6.
Strasser, A, O'Connor, L and Dixit, VM (2000). "Apoptosis signaling." Annu Rev Biochem 69: 217-45.
Susin, SA, Lorenzo, HK, Zamzami, N, Marzo, I, Snow, BE, Brothers, GM, Mangion, J, Jacotot, E, Costantini, P, Loeffler, M, Larochette, N, Goodlett, DR, Aebersold, R, Siderovski, DP, Penninger, JM and Kroemer, G (1999). "Molecular characterization of mitochondrial apoptosis-inducing factor." Nature 397(6718): 441-6.
Suzuki, Y, Nakabayashi, Y, Nakata, K, Reed, JC and Takahashi, R (2001). "X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes." J Biol Chem 276(29): 27058-63.
Takahashi, R, Deveraux, Q, Tamm, I, Welsh, K, Assa-Munt, N, Salvesen, GS and Reed, JC (1998). "A single BIR domain of XIAP sufficient for inhibiting caspases." J Biol Chem 273(14): 7787-90.
Tittel, JN and Steller, H (2000). "A comparison of programmed cell death between species." Genome Biol 1(3): REVIEWS0003.
Tsujimoto, Y and Shimizu, S (2000). "VDAC regulation by the Bcl-2 family of proteins." Cell Death Differ 7(12): 1174-81.
Van Cruchten, S and Van Den Broeck, W (2002). "Morphological and biochemical aspects of apoptosis, oncosis and necrosis." Anat Histol Embryol 31(4): 214-23.
van Loo, G, Saelens, X, van Gurp, M, MacFarlane, M, Martin, SJ and Vandenabeele, P (2002). "The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet." Cell Death Differ 9(10): 1031-42.
Varfolomeev, EE, Schuchmann, M, Luria, V, Chiannilkulchai, N, Beckmann, JS, Mett, IL, Rebrikov, D, Brodianski, VM, Kemper, OC, Kollet, O, Lapidot, T, Soffer, D, Sobe, T, Avraham, KB, Goncharov, T, Holtmann, H, Lonai, P and Wallach, D (1998). "Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally." Immunity 9(2): 267-76.
Vaux, DL, Cory, S and Adams, JM (1988). "Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells." Nature 335(6189): 440-2.
Verhagen, AM, Ekert, PG, Pakusch, M, Silke, J, Connolly, LM, Reid, GE, Moritz, RL, Simpson, RJ and Vaux, DL (2000). "Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins." Cell 102(1): 43-53.
Verhagen, AM, Silke, J, Ekert, PG, Pakusch, M, Kaufmann, H, Connolly, LM, Day, CL, Tikoo, A, Burke, R, Wrobel, C, Moritz, RL, Simpson, RJ and Vaux, DL (2002). "HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins." J Biol Chem 277(1): 445-54.
Vousden, KH and Lu, X (2002). "Live or let die: the cell's response to p53." Nat Rev Cancer 2(8): 594-604.
Wang, S, Miura, M, Jung, YK, Zhu, H, Li, E and Yuan, J (1998). "Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE." Cell 92(4): 501-9.
Wang, X (2001). "The expanding role of mitochondria in apoptosis." Genes Dev 15(22): 2922-33.
Wang, XW (1999). "Role of p53 and apoptosis in carcinogenesis." Anticancer Res 19(6A): 4759-71.
Wei, MC, Zong, WX, Cheng, EH, Lindsten, T, Panoutsakopoulou, V, Ross, AJ, Roth, KA, MacGregor, GR, Thompson, CB and Korsmeyer, SJ (2001). "Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death." Science 292(5517): 727-30.
Weil, M, Jacobson, MD, Coles, HS, Davies, TJ, Gardner, RL, Raff, KD and Raff, MC (1996). "Constitutive expression of the machinery for programmed cell death." J Cell Biol 133(5): 1053-9.
Wu, Y, Mehew, JW, Heckman, CA, Arcinas, M and Boxer, LM (2001). "Negative regulation of bcl-2 expression by p53 in hematopoietic cells." Oncogene 20(2): 240-51.
Yamada, KM and Araki, M (2001). "Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis." J Cell Sci 114(Pt 13): 2375-82.
Yang, J, Liu, X, Bhalla, K, Kim, CN, Ibrado, AM, Cai, J, Peng, TI, Jones, DP and Wang, X (1997). "Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked." Science 275(5303): 1129-32.
Yang, Y, Fang, S, Jensen, JP, Weissman, AM and Ashwell, JD (2000). "Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli." Science 288(5467): 874-7.
Yin, C, Knudson, CM, Korsmeyer, SJ and Van Dyke, T (1997). "Bax suppresses tumorigenesis and stimulates apoptosis in vivo." Nature 385(6617): 637-40.
Zuzarte-Luis, V and Hurle, JM (2002). "Programmed cell death in the developing limb." Int J Dev Biol 46(7): 871-6.